Authors: Robert M. Plenge, Edward M. Scolnick & David Altshuler
Synopsis
This review paper describes the potential of human genetics to perform target validation for drug development. The benefit of human genetics is that it directly involves interventions on the target of interest in humans, while no other preclinical model system can do this. The authors provide some examples of how human genetics informed target validation and discuss criteria for using genetics to prioritize drug targets.
Summary
Background
The drug development industry is notoriously inefficient. Less than 5% (!!) of targets that enter Phase 1 clinical trials are eventually approved by the FDA. The cost of drug development is dominated by cost of failures. Most failures are in Phase 2 clinical trials, and most drugs (50%) fail due to lack of efficacy (as opposed to toxicity, which is responsible for 25% of failures). These failures occur despite that fact every drug that enters clinical trials has shown evidence of efficacy and safety in preclinical models (cell line, organoid, and/or mouse/primate model organism). The issue lies with preclinical models not predicting benefit in patients.
The authors state that the primary goal of a preclinical model is to predict a dose-response curve between target perturbation and both efficacy and toxicity in humans. The most direct way to generate this curve is to perform an intervention on a target in humans. This can be accomplished in 3 ways:
-
Clinical trials. This is self-explanatory. For “me too” drugs (drugs with a similar structure or mechanism of action as other approved drugs), this information will be available.
- Naturally occurring environmental perturbation. An example is with steroids and rheumatoid arthritis (RA). In the 1930s, the following observations were made:
- Patients with RA had their symptoms improve both during pregnancy and following temporary stress brought upon by surgery. It was also known that pregnancy and stress resulted in higher levels of steroid hormones. The short time period between onset of pregnancy/stress and alleviation of RA symptoms helped establish causality between the two (according to the authors).
- Patients with Addison’s disease (adrenal insufficiency resulting in decreased steroid levels) had similar clinical features as RA patients
- Corticosteroids had anti-inflammatory activity in animal models
Based on these observations, it was hypothesized that cortisol would treat RA, which ended up being effective. One thing to note in the observations above that the target was modulated in both directions, which enabled a sort of dose-response curve to be generated. Adverse effects from excess steroids (diabetes, weight gain, etc.) could also be learned.
- Genetic perturbation. A prime example is LDL cholesterol and coronary artery disease. The following observations were made:
- There was an observational correlation between blood cholesterol and CAD in human populations
- Families with rare familial hypercholesterolaemia have a mutation in the LDL receptor (LDLR) gene, leading to high levels of LDL cholesterol and CAD
- Individual homozygous for the LDLR mutant were more severely affected than heterozygous individuals (implying some dose response)
Based on the above observations, statins targeting HMG-CoA reductase (HMGCR, known to be the rate-limiting enzyme in the cholesterol biosynthetic pathway) were hypothesized (and later shown) to be effective in treating LDL. Additional evidence for the LDL -> CAD hypothesis include the following subsequent developments:
- Families with rare gain-of-function mutations in PCSK9 had a higher incidence of CAD, while low-frequency LoF variants in PCSK9 were associated with reduced LDL and reduced CAD in GWAS. Note that again there is a bidirectional modulation of the target
- Animal studies showed PCSK9 was involved in post-translational regulation of LDLR. PCSK9-specific monoclonal antibodies were shown to reduce LDL cholesterol in clinical trials.
- A common non-coding variant in HMGCR was found to influence LDL and CAD in GWAS
- MR analysis showed a causal relationship between LDL and CAD. Meanwhile, MR analysis showed no relationship between HDL and CAD, which retrospectively supported clinical trials showing that raising HDL had no effect on CAD (It had previously been hypothesized that HDL also caused CAD based on observational correlations between HDL and CAD)
Table 1
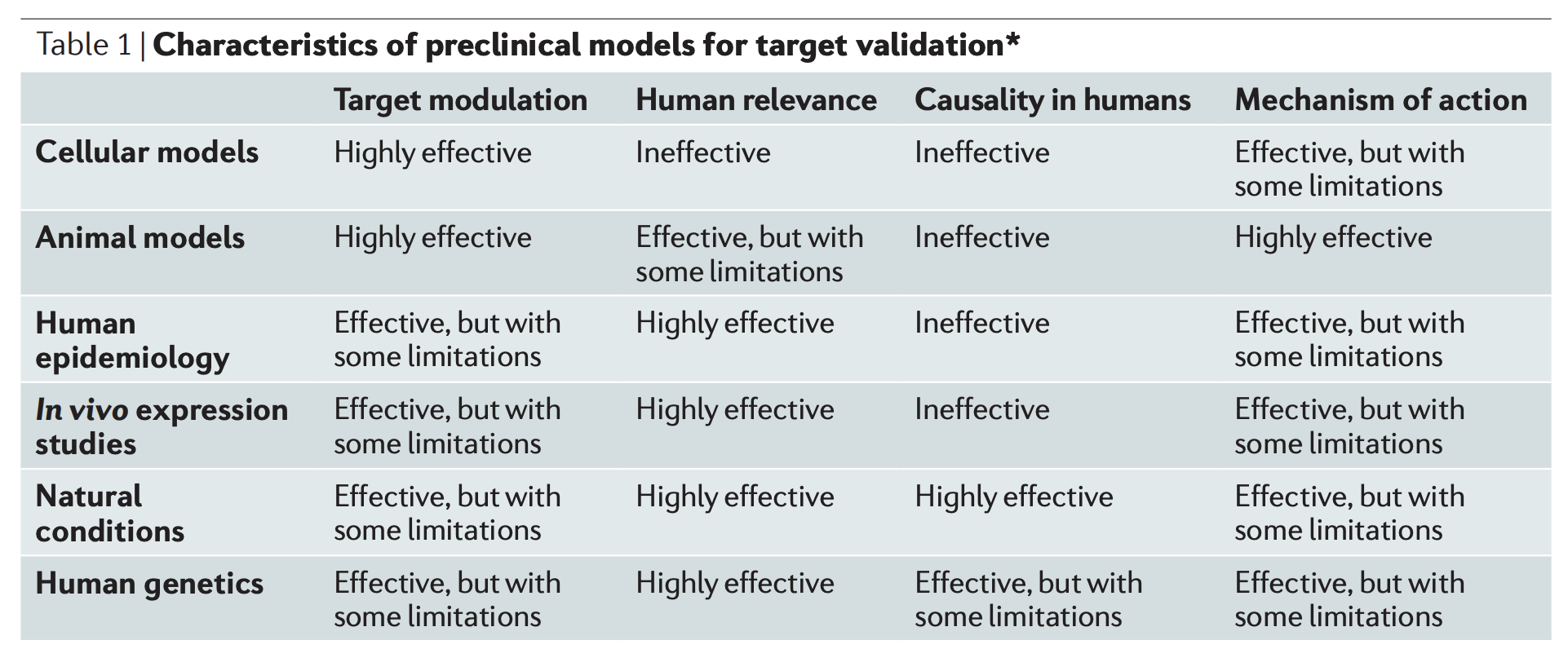
Aside from directly modulating the target in humans, other preclinical models include the above table. Here, “target modulation” refers to the ability to modulate the target, while “mechanism of action” refers to the ability to understand the relationship between the biological mechanism of the model and human disease. I don’t understand why many of the labels here are chosen (the authors don’t justify most of them), so here’s my interpretation of the table:
- Cellular models: It’s straightforward to modulate the target of interest, but there is a big gap in complexity between the model and human biology
- Animal models: It’s straightforward to modulate the target of interest and the gap to humans is smaller than cellular models, but still large. I think the author’s claim that “mechanism of action” is most effective for animal models stems from the fact that animals can be tested invasively/dissected, whereas humans cannot.
- Human epidemiology: Only observational correlations can be observed, so results are far from causality. However, epidemiology can be used to identify potential targets and rule out others. Human expression studies are a subset of this category.
- Natural conditions: I don’t understand why “causality in humans” is labelled highly effective for this category; it seems like it would be hard to know whether a natural condition is independent of confounders (whereas for human genetics, randomization is obtained via meiosis and random mating)
- Human genetics: Perhaps gets the closest to causality in humans without actually performing a clinical trial. However, targets are selected randomly by random mutations and are subjected to the forces of natural selection (which removes likely disease-relevant variants with large effects on fitness). Moreover, establishing a link between the genetic variant and target/molecular process is often very non-trivial.
Box 2
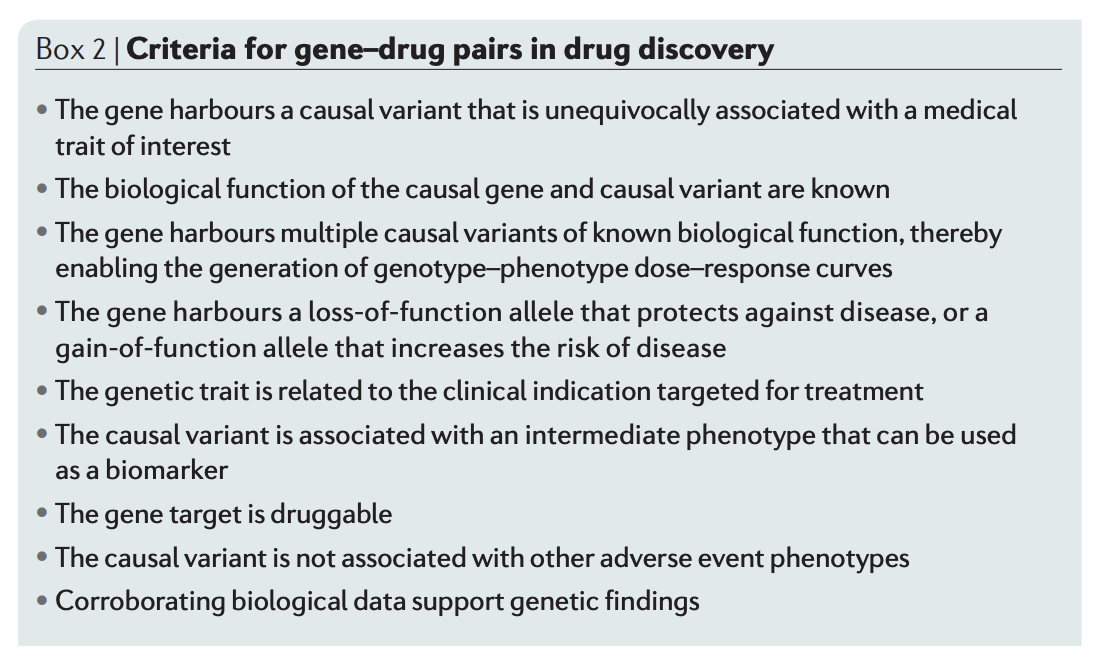
Here, the authors give some recommendations for selecting targets based on human genetic evidence. Some interesting criteria to note:
- Gene should have LOF allele that protects against disease or GOF allele that increases disease. Apparently it’s easier to develop drugs that are inhibitors rather than activators.
- Variant is associated with intermediate phenotype that can be used as a biomarker. Biomarkers for disease are important since they can be used as surrogate endpoints in clinical trials, enable easier enrollment and tracking disease progress, etc.
Table 2
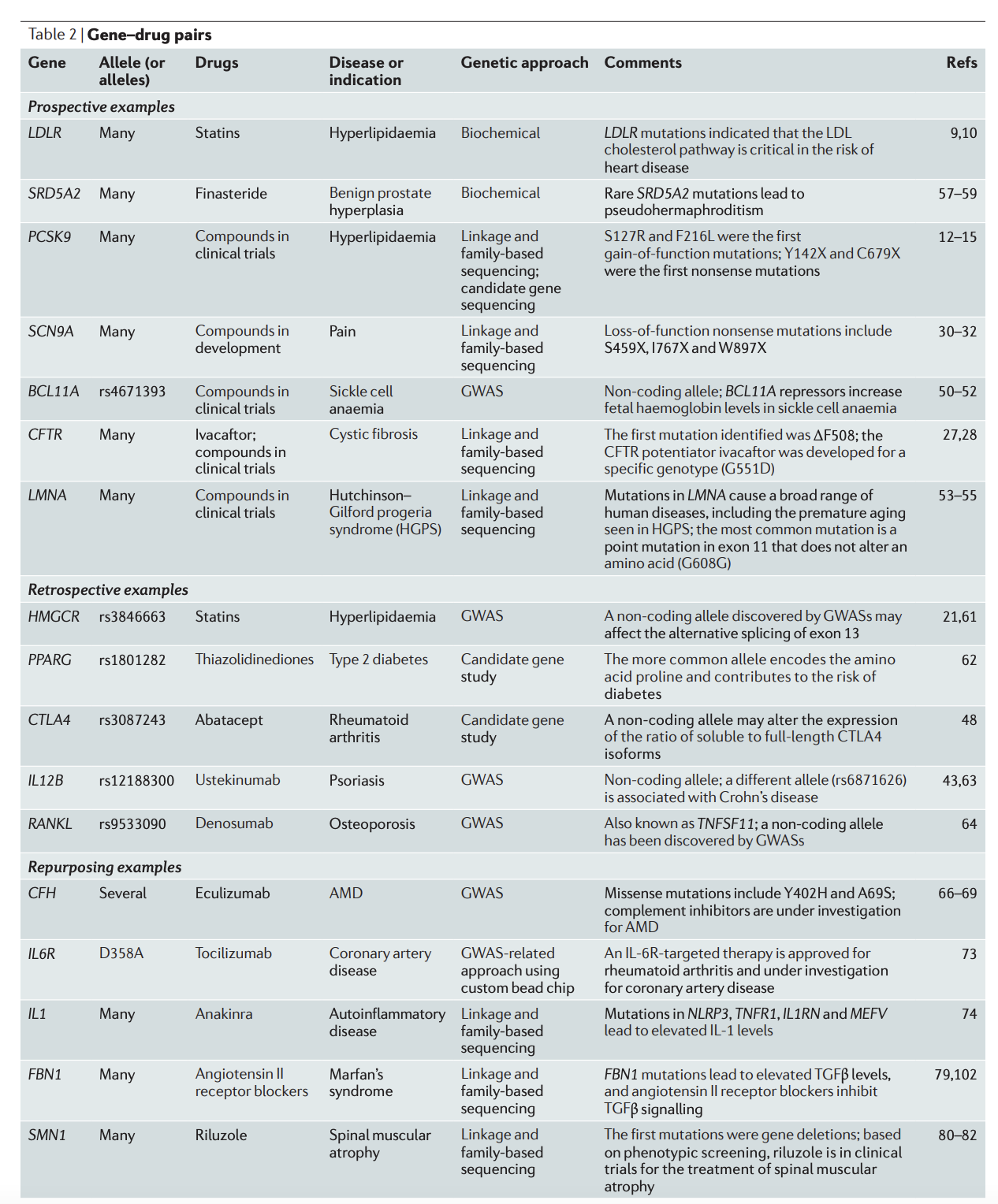
This table shows historical examples of genetics for target validation. For prospective examples, genetics played an important role in the development of the drug. For retrospective examples, genetics didn’t play a large role in drug development, but was later shown to identify the drug target. For repurposing examples, genetics contributed toward repurposing an existing drug for a different indication. One thing to note is that BCL11A for sickle cell anemia was the only example here in which GWAS directly led to a drug being developed.
Drawbacks of genetics to prioritize drug targets
- Gene must by chance have a series of alleles (from common to rare) that modulates its activity in a way that can be mimicked by a drug
- Many safe and effective drugs have been developed without any genetic evidence
- Biological mechanisms that cause disease may not be the same that should be targeted to treat disease. For example, the immune system is involved in the cause of T1D, but once the pancreas is destroyed treatment involves just administering insulin.
- Complex diseases have many 100s or 1000s of “causal” genes
- Notion of targeting single genes is reductionist; actual biology (especially for complex diseases) is much more complicated