Research interests
I am broadly interested in anything to do with statistics/machine learning, biotechnology, and drug development. As an undergraduate, I conducted research in computational cancer genomics. I conceived and led a project aiming to identify associations between gene expression levels and a cancer phenotype known as “genomic instability” (which is a metric of how quickly tumors mutate) in large databases of tumor sequencing data. The project culminated in a manuscript I wrote that was published in Scientific Reports.
As a graduate student, my research has broadly been in genetics. Specifically, two subareas: statistical genetics and functional genomics technology development. Statistical genetics is a field dating back to Ronald Fisher in the 1920s which aims to study the statistical relationship between genetics and phenotype in human or animal populations. In this area, I worked on a project that involved defining a quantity known as “heritability mediated by gene expression levels,” which quantifies how much total genetic effect on a phenotype “flows through” separately measured gene expression levels. I derived a novel method of moments estimator of this quantity, established its properties (including bias, consistency, and calibration under the null) via derivations and simulations, and applied it to real human disease genetics data. The project culminated in a manuscript I wrote that was published in Nature Genetics.
Functional genomics technology development is a field that involves developing new experimental assays to explore the functional relationship between genetics and phenotype in a high-throughput fashion. In this area, I worked on a project that developed a new type of Perturb-seq assay. Perturb-seq is a type of genetic screen that involves making many targeted perturbations using CRISPR to cells, then reading out the effects of the perturbations on the transcriptome using single-cell RNA-sequencing. Prior to my work, this assay was prohibitively expensive to run, costing millions of dollars for genome-wide screens. Using new computational and experimental strategies inspired by compressed sensing, I helped reduce the cost of the experiment by an order of magnitude, making it much more feasible to run for many labs. The project culminated in a manuscript I wrote that was published in Nature Biotechnology.
For more info on my past work, see below.
Current research
I am currently interested in thinking about ways to develop drugs efficiently.
Past research
Functional genomics technology development
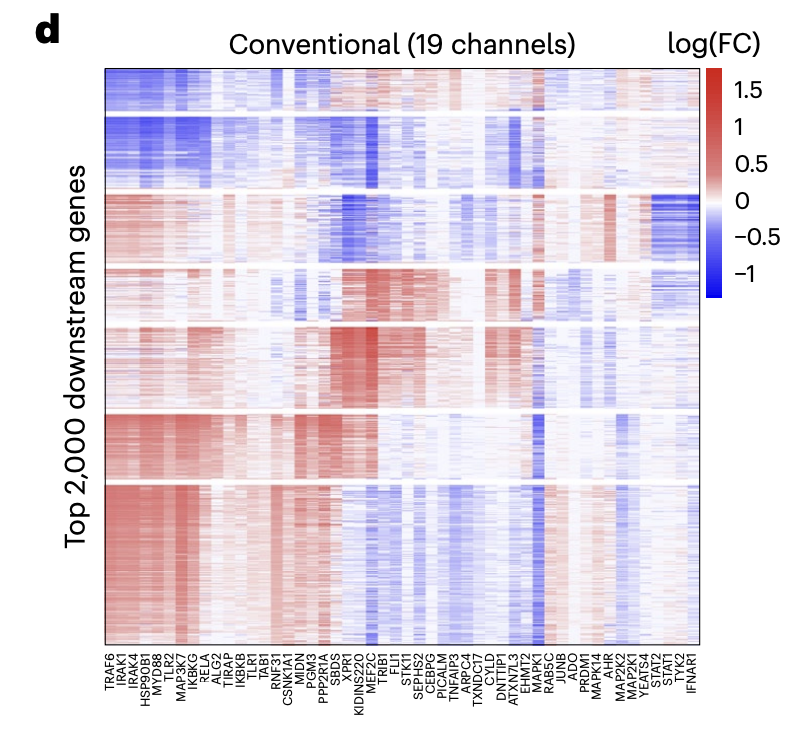
Douglas Yao, Loic Binan, Jon Bezney, Brooke Simonton, Jahanara Freedman, Chris Frangieh, Kushal Dey, Kathryn Geiger-Schuller, Basak Eraslan, Alexander Gusev, Aviv Regev, Brian Cleary. Scalable genetic screening for regulatory circuits using Compressed Perturb-seq. Nature Biotechnology, 2023.
See here for a summary of the work.
Statistical genetics
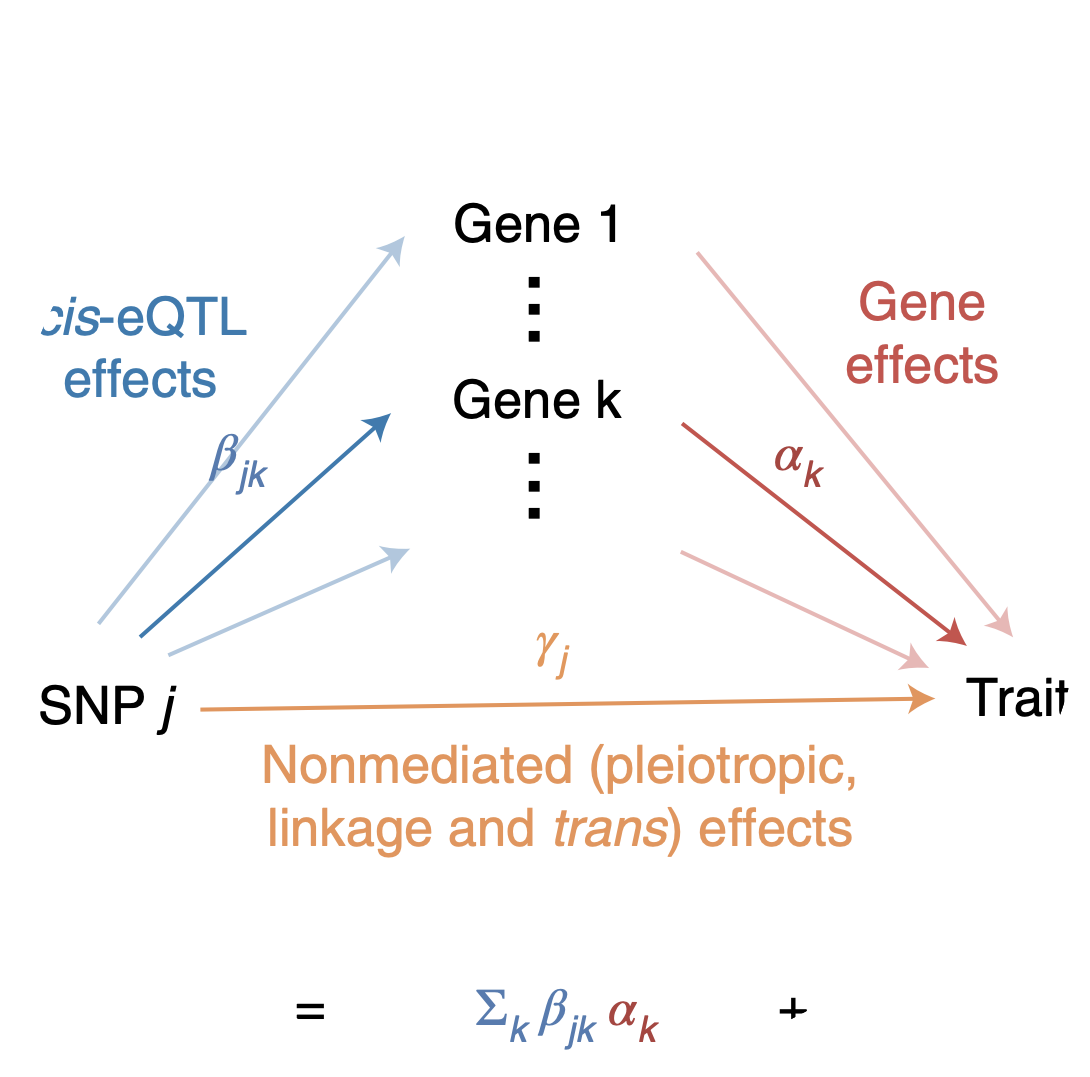
Douglas Yao, Luke O’Connor, Alkes Price, Alexander Gusev. Quantifying genetic effects on disease mediated by assayed gene expression levels. Nature Genetics, 2020.
See here for a summary of the work aimed at a general audience, and here for a summary of the work aimed at a technical audience.
Computational cancer genomics
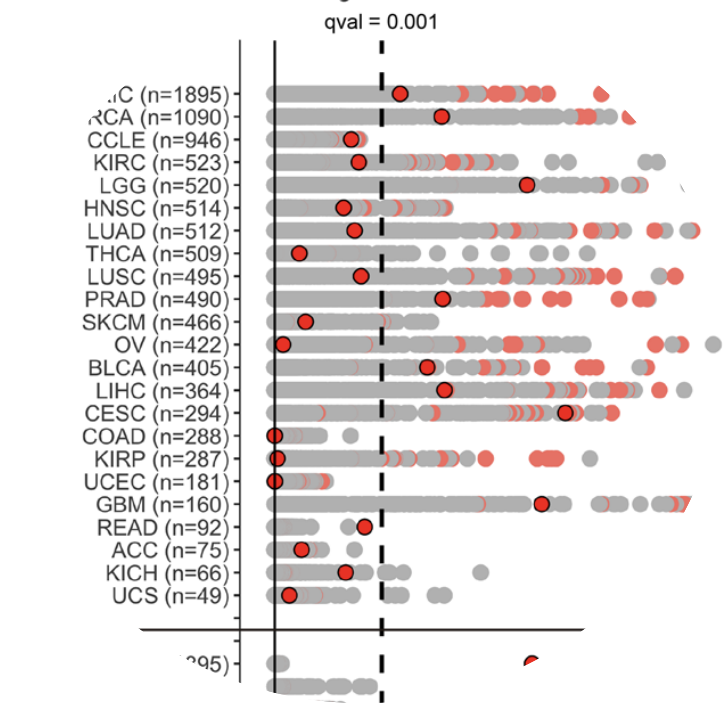
Douglas Yao, Nikolas Balanis, Eleazar Eskin, Thomas Graeber. A linear mixed model approach to gene expression-tumor aneuploidy association studies. Scientific Reports, 2019.
Other publications
Katherine Siewert-Rocks, Samuel Kim, Douglas Yao, Huwenbo Shi, Alkes Price. Leveraging gene co-regulation to identify gene sets enriched for disease heritability. American Journal of Human Genetics, 2022.
Igor Mandric, Harry Taegyun Yang, Nicolas Strauli, Dennis Montoya, … , Douglas Yao, … , et al. Profiling immunoglobulin repertoires across multiple human tissues by RNA sequencing. Nature Communications, 2020.
Keith Mitchell, Jaqueline Brito, Igor Mandric, Qiaozhen Wu, … , Douglas Yao, … , et al. Benchmarking of computational error-correction methods for next-generation sequencing data. Genome Biology, 2020.